

Raquitismo hipofosfatémico: ¿Qué es?
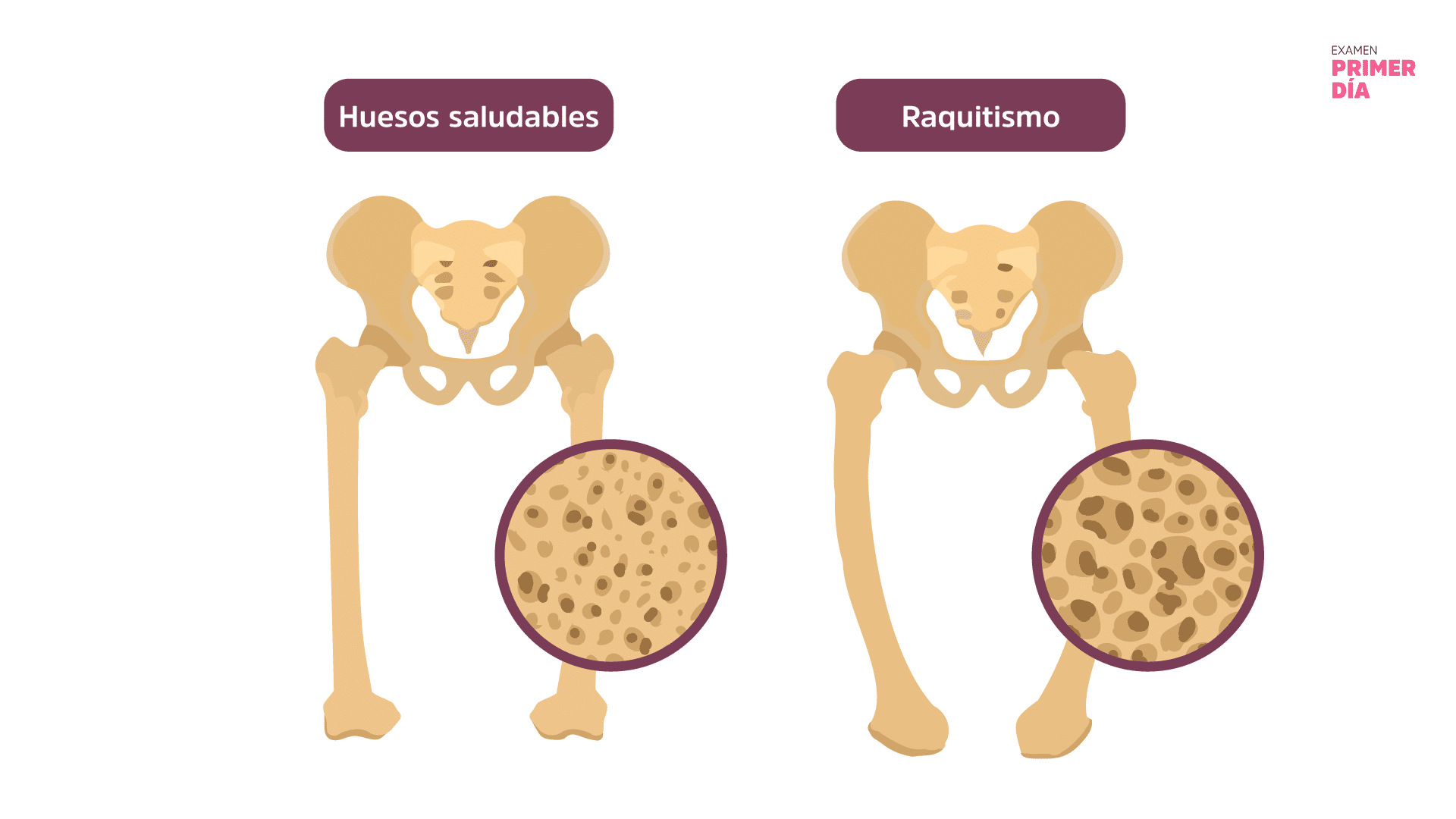
El raquitismo hipofosfatémico es una enfermedad rara y progresiva causada por una insuficiencia renal que provoca niveles bajos de fosfato en la sangre (hipofosfatemia), lo que hace que los huesos sean dolorosamente blandos y se curven fácilmente.
Si no se trata, especialmente durante el desarrollo óseo del niño, provoca cambios irreversibles en los huesos y los dientes. Sin embargo, tras un tratamiento adecuado, los niños afectados por la enfermedad pueden tener una vida larga y saludable.
¿Qué es el Raquitismo?El raquitismo es una enfermedad ósea de la infancia que se caracteriza por un defecto en la mineralización ósea que da lugar a anomalías en el cartílago de la placa de crecimiento, que se observan sobre todo en los huesos largos. Puede producirse por una carencia de calcio, fósforo o vitamina D. El raquitismo hipofosfatémico fue descrito por primera vez como raquitismo resistente a la vitamina D por Fuller Albright, porque un paciente no respondió al tratamiento con vitamina D. En aquella época, la deficiencia de vitamina D era la causa más común de raquitismo. En el caso del raquitismo hipofosfatémico, este es causado por mutaciones en los genes que intervienen en la regulación del fosfato. |
Conozca mejor el panel de enfermedades esqueléticas que Mendelics ofrece.
¿Cuáles son los síntomas del raquitismo hipofosfatémico?
Los síntomas aparecen en los primeros años de vida, haciéndose más evidentes después de que el niño empiece a caminar, y pueden variar en el nivel de gravedad, incluso entre los miembros afectados de una misma familia.
Las formas graves pueden causar dolor en los huesos y las articulaciones, desarrollando huesos frágiles, arqueo de las piernas y otras deformidades óseas y de baja estatura. Algunos bebés afectados pueden presentar craneosinostosis, es decir, el cierre precoz de las suturas craneales, que puede afectar al desarrollo del niño.
Como es una enfermedad progresiva, si no se trata, los síntomas empeoran con el tiempo.
¿Qué causa el raquitismo hipofosfatémico?
El raquitismo hipofosfatémico es casi siempre genético y hereditario, es decir, causado por mutaciones heredadas de los padres.
En raras ocasiones, la enfermedad se desarrolla como resultado de ciertos tipos de cáncer, como tumores de células gigantes del hueso, los sarcomas, el cáncer de próstata y el cáncer de mama.
La enfermedad se produce debido a un desequilibrio del fosfato en el organismo que, entre sus muchas funciones, desempeña un papel fundamental en la formación y el crecimiento de los huesos en la infancia y ayuda a mantener la resistencia ósea en los adultos.
Normalmente, los niveles de fosfato en el organismo están controlados en gran medida por los riñones: cuando hay en exceso se excreta en la orina y cuando sus niveles son bajos es reabsorbido.
Sin embargo, en las personas con raquitismo hipofosfatémico, los riñones no llevan a cabo la reabsorción de fosfato de manera eficiente, lo que hace que sea eliminado por la orina más de lo necesario. Como resultado, no hay suficiente fosfato en el torrente sanguíneo para que los huesos se desarrollen y se mantengan adecuadamente.
El raquitismo hipofosfatémico puede estar causado por mutaciones en varios genes que, en general, regulan directa o indirectamente una proteína que normalmente inhibe la capacidad de los riñones para reabsorber el fosfato en la sangre.
El gen implicado determina el tipo de raquitismo hipofosfatémico y cómo se hereda:
- El tipo más común es el raquitismo hipofosfatémico ligado al cromosoma X, causado por una mutación en el gen PHEX.
- Otros genes que pueden ser responsables de la enfermedad son: CLCN5, DMP1, ENPP1, FGF23 y SLC34A3 y causan la enfermedad siguiendo otros patrones de herencia: recesivo ligado al cromosoma X, autosómico dominante o autosómico recesivo.
Raquitismo Hipofosfatémico ligado al cromosoma XEl raquitismo hipofosfatémico ligado al cromosoma X (XLH) afecta aproximadamente a 1 de cada 25.000 nacidos vivos y se desarrolla en los primeros años de vida. Dado que los síntomas son similares, los pacientes suelen ser diagnosticados erróneamente con deficiencia de vitamina D, lo que retrasa su diagnóstico y, en consecuencia, el inicio del tratamiento. El raquitismo hipofosfatémico ligado al cromosoma X es el resultado de mutaciones en el gen PHEX, y es heredado siguiendo un patrón dominante ligado al X (hipofosfatemia ligada al X), por lo que tanto en las mujeres (que tienen dos cromosomas X) como en los hombres (que sólo tienen un cromosoma X), una copia alterada del gen es suficiente para causar la enfermedad. Las mujeres afectadas tienen un 50% de posibilidades de transmitir el gen mutado PHEX a sus hijos, independientemente del sexo. Los hombres afectados, en cambio, transmitirán el gen mutado a todas sus hijas y a ninguno de sus hijos varones, porque los niños sólo reciben el cromosoma Y de sus padres. Las mutaciones en el gen PHEX también pueden causar la enfermedad con el patrón de herencia recesivo ligado al cromosoma X, conocido como enfermedad de Dent. En este caso, una copia alterada del gen es suficiente para causar la enfermedad en los hombres, pero es necesario que ambas copias (materna y paterna) estén alteradas para que la enfermedad se manifieste en las mujeres. El 23 de junio es el Día de la Concientización sobre el Raquitismo Hipofosfatémico Ligado al X. Conocido como #XLHDay, la campaña pretende concientizar a las personas sobre la enfermedad y la importancia del diagnóstico precoz.
|

¿Cuál es el tratamiento del raquitismo hipofosfatémico?
El tratamiento del raquitismo hipofosfatémico depende de la causa subyacente y puede ser clínico, farmacológico y quirúrgico.
En Brasil, desde 2022, el tratamiento de los pacientes con raquitismo hipofosfatémico es apoyado por el protocolo clínico y de directrices terapéuticas (PCDT) y proporcionado por el Sistema Único de Salud (SUS).
El tratamiento convencional para los pacientes con XLH se realizaba con fosfato y vitamina D, que no tratan directamente la causa de esta enfermedad. Ahora, pueden recibir el tratamiento con el fármaco de referencia, el burosumab: un anticuerpo monoclonal que actúa sobre las proteínas asociadas a la enfermedad, aumentando la reabsorción de fosfato del riñón y reduciendo el daño causado por la XLH.
Raquitismo hipofosfatémico: ¿Cómo se hace el diagnóstico?
Los signos y síntomas clínicos, las radiografías óseas con hallazgos sugestivos y las pruebas bioquímicas alteradas indican una sospecha de la enfermedad, que debe ser confirmada por exámenes genéticos, mediante la identificación de mutaciones en el gen PHEX, principalmente.
¿Por qué es importante el Examen Primer Día para el diagnóstico precoz del raquitismo hipofosfatémico?
La XLH es una de las más de 340 enfermedades examinadas por el Examen Primer Día, el primer examen de tamizaje genético neonatal para recién nacidos en Brasil y que ahora está disponible en toda Latinoamérica, y el único examen que detecta el raquitismo hipofosfatémico ligado al cromosoma X.
Este examen genético analiza directamente el ADN del bebé y, por tanto, es capaz de identificar qué mutación del gen PHEX causa la enfermedad.
El examen Primer Día puede realizarse desde el nacimiento del bebé, antes de la aparición de cualquier indicio clínico de la enfermedad. Además, la toma de muestra es muy sencilla, se hace con un hisopo.
Se sospecha que mi hijo tiene raquitismo hipofosfatémico, ¿Puedo hacer la prueba del Examen Primer Día para confirmar el diagnóstico?
El Examen Primer Día es una examen de Tamizaje Neonatal Genético que pretende identificar a los bebés con riesgo de desarrollar enfermedades que se manifiestan durante los primeros meses o años de vida.
Cuando el niño ya manifiesta signos de alguna enfermedad, se recomienda realizar un examen de diagnóstico genético para confirmar la sospecha. Para el diagnóstico de XLH, Mendelics ofrece exámenes para confirmar el diagnóstico de Raquitismo Hipofosfatémico, incluyendo el Panel de Enfermedades Tratables y el Panel de Enfermedades Esqueléticas.
Es importante tener en cuenta que los exámenes de diagnóstico, a diferencia de las pruebas de tamizaje, sólo pueden realizarse a petición del médico y con seguimiento médico.
Hable con su médico y póngase en contacto con nuestro equipo para obtener más información.
Referencias
- “Familial hypophosphatemia,” NORD (National Organization for Rare Disorders). Acesso em 28 de maio de 2021.
- Tavana N, Thilakavathy K, Kennerson ML, Ting TH. Genetic basis of hereditary hypophosphataemic rickets and phenotype presentation in children and adults. Endokrynol Pol. 2021;72(4):366-394.
- “Learn About XLH ”, XLH Network. Acesso em 28 de maio de 2021.
- D. Haffner et al., “Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia,” Nature Reviews Nephrology, vol. 15, no. 7, pp. 435–455, May 2019.
- Projeto de Lei n 397, de 2020, Assembléia Legislativa do Estado de São Paulo. https://www.al.sp.gov.br/propositura/?id=1000327611
0 comentários